
Atavide: importing your data into R
This post is one of a multipart series about analysing your metagenomics samples with Atavide. It is intended to be a quick and dirty look at the superfocus output, how you can make it better, importing into R, and how you might go about some simple plots and tests.
Dependencies
For linux, you can install taxonkit from bioconda. Follow the documentation for installing or downloading the database.
conda install -c bioconda taxonkit
Install dependencies for R (Rstudio). You can do everything in base R, but these packages make things easier.
install.packages("dplyr")
install.packages("tidyr")
install.packages("ggplot2")
library(dplyr)
library(tidyr)
library(ggplot2)
ReadAnnotations/all_taxonomy.tsv (Superfocus output)
Look at the file (in Linux)
head all_taxonomy.tsv
Superkingdom|Phylum|Class|Order|Family|Genus|Species|Taxid FAME000065 FAME000066 FAME000067 FAME000068 FAME000069 FAME000070 FAME000072 FAME000073 FAME000074 FAME000075 FAME000076 FAME000077 FAME000078 FAME000079 FAME000080 FAME000081 FAME000082
s:Archaea|p:Candidatus Korarchaeota|c:|o:|f:|g:Candidatus Korarchaeum|s:Candidatus Korarchaeum cryptofilum|taxid: 374847 1 0 0 1 2 0 0 1 10 0 0 0 0 2 0 0
s:Archaea|p:Crenarchaeota|c:Thermoprotei|o:Desulfurococcales|f:Desulfurococcaceae|g:Aeropyrum|s:Aeropyrum pernix|taxid: 272557 0 0 0 0 0 0 0 0 00 0 0 0 0 1 0 0
s:Archaea|p:Crenarchaeota|c:Thermoprotei|o:Desulfurococcales|f:Desulfurococcaceae|g:Ignicoccus|s:Ignicoccus hospitalis|taxid: 453591 0 0 0 2 0 0 0 00 2 0 0 0 0 0 0 0
s:Archaea|p:Crenarchaeota|c:Thermoprotei|o:Desulfurococcales|f:Desulfurococcaceae|g:Staphylothermus|s:Staphylothermus marinus|taxid: 399550 1 0 0 0 0 1 00 1 2 0 0 0 0 0 0 0
s:Archaea|p:Crenarchaeota|c:Thermoprotei|o:Desulfurococcales|f:Pyrodictiaceae|g:Hyperthermus|s:Hyperthermus butylicus|taxid: 415426 0 1 0 1 0 0 0 00 0 0 0 0 0 0 0 0
s:Archaea|p:Crenarchaeota|c:Thermoprotei|o:Sulfolobales|f:Sulfolobaceae|g:Sulfolobus|s:Sulfolobus acidocaldarius|taxid: 330779 0 0 0 0 0 0 0 0 00 0 1 0 0 0 1 0
s:Archaea|p:Crenarchaeota|c:Thermoprotei|o:Sulfolobales|f:Sulfolobaceae|g:Sulfurisphaera|s:Sulfurisphaera tokodaii|taxid: 273063 0 0 0 0 0 0 1 00 0 0 0 0 0 0 0 0
s:Archaea|p:Crenarchaeota|c:Thermoprotei|o:Thermoproteales|f:Thermofilaceae|g:Thermofilum|s:Thermofilum pendens|taxid: 368408 0 1 1 0 2 1 2 2 00 0 0 0 0 0 1 0
s:Archaea|p:Crenarchaeota|c:Thermoprotei|o:Thermoproteales|f:Thermoproteaceae|g:Caldivirga|s:Caldivirga maquilingensis|taxid: 397948 2 0 0 0 0 0 0 00 0 0 0 0 0 0 0 2
tail all_taxonomy.tsv
s:|p:Uroviricota|c:Caudoviricetes|o:Caudovirales|f:Siphoviridae|g:|s:Streptococcus phage phi3396|taxid: 423476 0 4 0 25 228 6 3 65 0 7 10 0 11 0 6 0
s:|p:Uroviricota|c:Caudoviricetes|o:Caudovirales|f:Siphoviridae|g:|s:Streptomyces phage VWB|taxid: 10702 0 2 1 30 146 0 0 0 0 0 80 0 0 0 0 36 0
s:|p:Uroviricota|c:Caudoviricetes|o:Caudovirales|f:Siphoviridae|g:|s:Streptomyces phage mu1/6|taxid: 370623 0 0 0 1 0 0 0 0 0 0 00 1 0 0 0 0
s:|p:Uroviricota|c:Caudoviricetes|o:Caudovirales|f:Siphoviridae|g:|s:Temperate phage phiNIH1.1|taxid: 173707 9 105 25 252 404 163 40 915 1 170 90 18 0 0 67 8
s:|p:Uroviricota|c:Caudoviricetes|o:Caudovirales|f:Siphoviridae|g:|s:Yersinia phage PY54|taxid: 172667 0 0 0 1 1 0 1 0 0 2 0 00 1 0 0 1
s:|p:Uroviricota|c:Caudoviricetes|o:Caudovirales|f:|g:|s:Enterobacteria phage P4|taxid: 10680 1 3 0 0 0 0 1 0 2 0 0 0 10 0 1 0
s:|p:Uroviricota|c:Caudoviricetes|o:Caudovirales|f:|g:|s:Streptococcus phage 315.5|taxid: 198542 6 19 123 60 159 8 1 14 70 82 4 15 0 1 0 9 1
s:|p:|c:|o:|f:|g:|s:Phage Gifsy-1|taxid: 129861 0 0 0 0 1 0 0 0 0 0 0 0 1 0 0 0 0
s:|p:|c:|o:|f:|g:|s:Phage Gifsy-2|taxid: 129862 0 0 0 0 0 0 0 0 0 8 0 0 0 0 1 0 0
s:|p:|c:|o:|f:|g:|s:Salmonella phage Fels-1|taxid: 128975
ewww.
It’s a weird format table but the first thing we need to fix is the missing taxonomic information. We use Taxonkit for this.
sed 's/.*taxid: //' ReadAnnotations/all_taxonomy.tsv \
| taxonkit lineage -i 1 --data-dir ~/data/git/hecatomb/databases/tax/taxonomy/ \
| taxonkit reformat --data-dir ~/data/git/hecatomb/databases/tax/taxonomy/ -i 19 -f "{k}\t{p}\t{c}\t{o}\t{f}\t{g}\t{s}" -F --fill-miss-rank \
| cut --complement -f1,19 \
> all_tax_fixed.tsv
There’s a lot going on here so lets break it down. Sed is stripping everything but the taxid number from the first column. taxonkit lineage is translating the taxid into a taxonomy (delimited with ‘;’) and adding it to the end of each line (column 19). taxonkit reformat is reading in the taxonomy (column 19), filling in any missing fields, and outputing in a tab separated format (columns 20 onwards). We dont need columns 1 (tax IDs) and 19 (default format taxonomy with missing fields), so we remove them using Cut.
For taxonkit, make sure you’ve either installed the databases, or downloaded them somewhere so you can point to them with --data-dir (or just use my folder on deepthought).
head all_tax_fixed.tsv
FAME000065 FAME000066 FAME000067 FAME000068 FAME000069 FAME000070 FAME000072 FAME000073 FAME000074 FAME000075 FAME000076 FAME000077 FAME000078 FAME000079 FAME000080 FAME000081 FAME000082
31 42 11 33 138 50 7 133 34 58 22 21 6 22 18 15 4 Archaea Candidatus Korarchaeota unclassified Candidatus Korarchaeota class unclassified Candidatus Korarchaeota order unclassified Candidatus Korarchaeota family Candidatus Korarchaeum Candidatus Korarchaeum cryptofilum
8 9 2 8 22 9 5 2 29 30 8 0 11 27 41 4 2 Archaea Crenarchaeota Thermoprotei Desulfurococcales Desulfurococcaceae Aeropyrum Aeropyrum pernix
2 9 0 0 2 0 0 1 3 5 2 0 1 2 7 2 0 Archaea Crenarchaeota Thermoprotei Desulfurococcales Desulfurococcaceae Desulfurococcus Desulfurococcus amylolyticus
11 3 8 20 38 11 3 11 13 29 4 3 0 12 8 7 14 Archaea Crenarchaeota Thermoprotei Desulfurococcales Desulfurococcaceae Ignicoccus Ignicoccus hospitalis
115 32 39 69 250 49 74 10 69 153 46 24 48 31 25 35 26 Archaea Crenarchaeota Thermoprotei Desulfurococcales Desulfurococcaceae Staphylothermus Staphylothermus marinus
60 15 16 31 74 20 23 6 53 58 26 3 30 29 8 6 10 Archaea Crenarchaeota Thermoprotei Desulfurococcales Pyrodictiaceae Hyperthermus Hyperthermus butylicus
36 3 12 23 50 11 7 15 33 30 7 14 3 5 9 9 17 Archaea Crenarchaeota Thermoprotei Sulfolobales Sulfolobaceae Metallosphaera Metallosphaera sedula
4 0 2 2 7 0 0 0 3 12 8 0 0 3 4 3 1 Archaea Crenarchaeota Thermoprotei Sulfolobales Sulfolobaceae Saccharolobus Saccharolobus solfataricus
3 0 6 8 20 6 0 1 1 21 2 5 1 27 11 56 4 Archaea Crenarchaeota Thermoprotei Sulfolobales Sulfolobaceae Sulfolobus Sulfolobus acidocaldarius
The file is missing the header information for the new taxonomy fields, so we can fix that before importing into R, or after importing into R. You can open it up in a text editor and do it manually, adding in tabs to separate the header fields. The last sample name is ‘FAME000082’ so I’ll just use sed.
sed 's/FAME000082\t/FAME000082\tKingdom\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies/' all_tax_fixed.tsv > all_tax_final.tsv
Read the file into R (assuming you’ve downloaded it)
data = read.csv("all_taxonomy.tsv",header=T,sep="\t")
View(data)
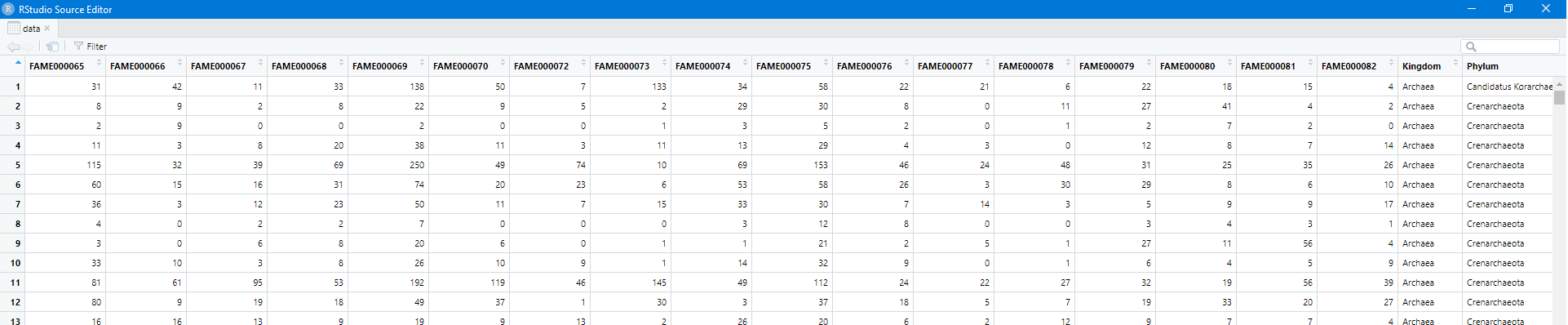
At the moment this is a wide table, and I prefer to work in long table format.
The samples are in columns 1 to 18 (R is 1-indexed).
We want to have a column for the tax information, a column for the sample, and a column for the number.
Use the tidyr function gather() to convert the table.
longData = gather(data, "sample", "count", 1:17)
View(longData)
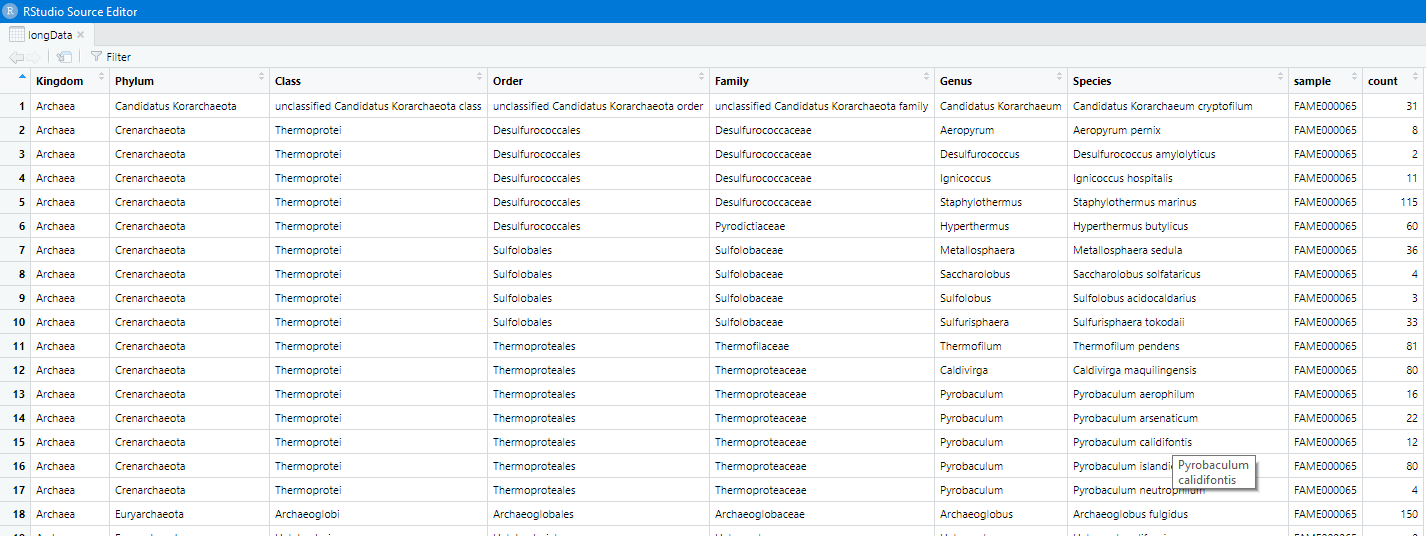
Let’s add some metadata, first add it in as a data frame, you can read it in from a file or past in the raw text. NOTE: R will read the group numbers in the table below into numerical values, which is fine until we go to plot. We want R to treat them as categories, so we need to coerce them into character strings.
meta = read.table(text = "sample,group
FAME000065,1
FAME000066,1
FAME000067,1
FAME000068,1
FAME000069,1
FAME000070,1
FAME000072,1
FAME000073,1
FAME000074,1
FAME000075,2
FAME000076,2
FAME000077,2
FAME000078,2
FAME000079,2
FAME000080,2
FAME000081,2
FAME000082,2",sep=',',header=T)
meta$group = as.character(meta$group)
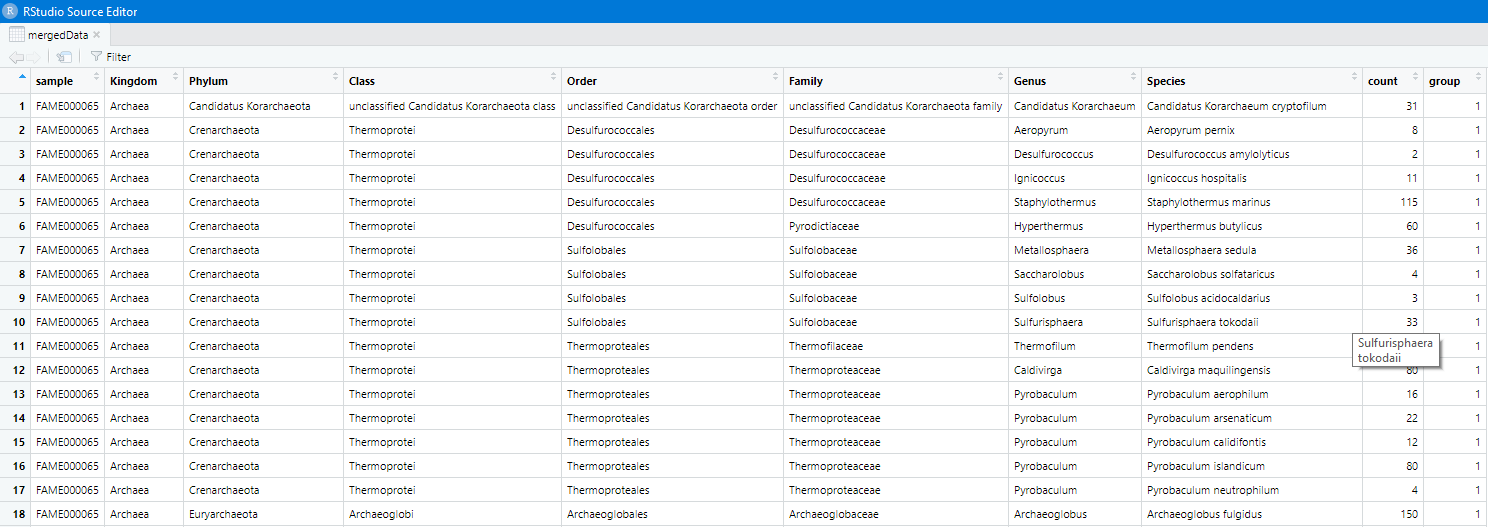
Merge it with our taxonomy table
mergedData = merge(longData, meta, by='sample')
View(mergedData)
Let’s plot the counts of each sample for each phyla, colored by their group.
ggplot(mergedData)+geom_point(aes(y=Phylum,x=count,color=group))

It looks like Firmicutes might be interesting, let’s just plot that for both groups.
The example below is a base-R method for slicing dataframes.
mergedData$Phylum=='Firmicutes' will return a list of True or False,
which can be used to specify which rows we want from mergedData.
firmSub = mergedData[mergedData$Phylum=='Firmicutes',]
ggplot(firmSub)+geom_boxplot(aes(x=group,y=count),width=0.1)+scale_y_log10()
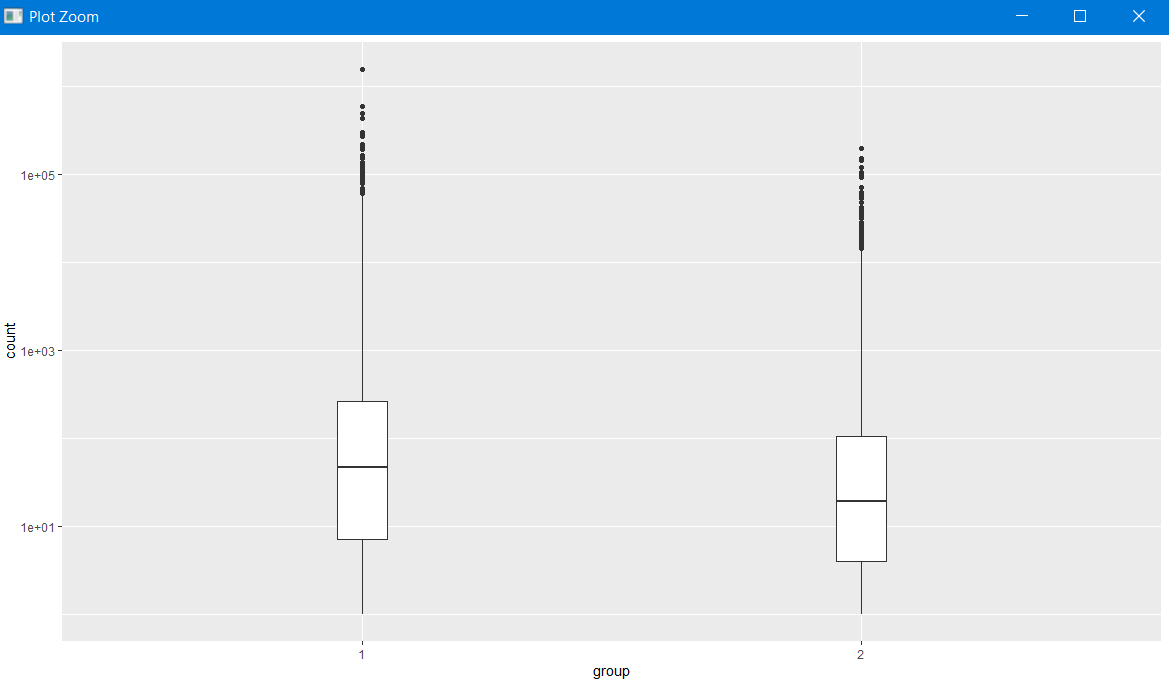
This shows one point per species, only showing the Firmicutes species. Let’s sum all the Firmicutes counts for each sample.
FirmSum = mergedData %>% dplyr::filter(Phylum == "Firmicutes") %>% group_by(sample) %>% summarise(sum(count))
FirmSum = merge(FirmSum, meta, by = 'sample')
colnames(FirmSum) = c('sample','count','group')
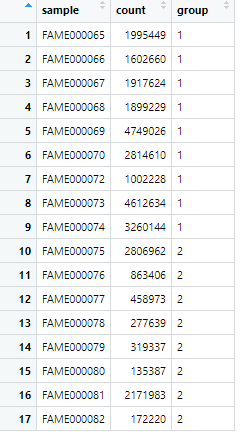
We refilter (to show the dplyr way to filter), group by sample, and use the summarise function to sum the counts for each sample. We then add the metadata back in for plotting and fix the header.
ggplot(FirmSum)+geom_jitter(aes(x=group,y=count,color=sample),width=0.1)
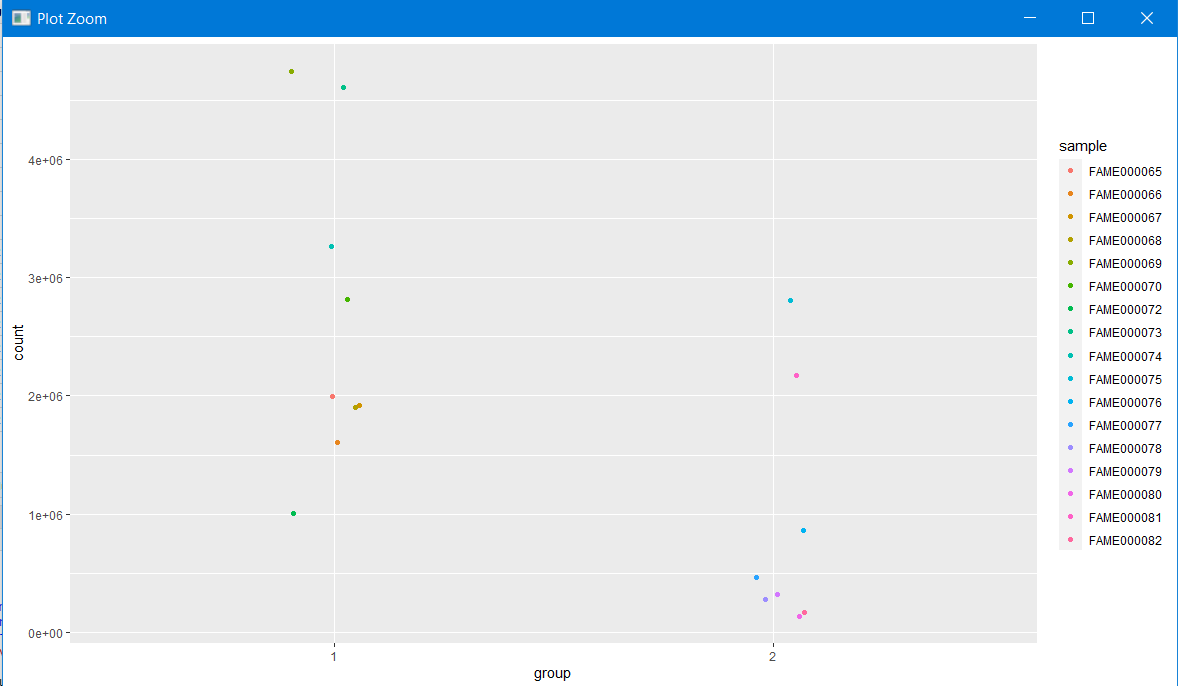
Is it significant? Lets do a simple t-test
t.test(FirmSum[FirmSum$group=='1',]$count, FirmSum[FirmSum$group=='2',]$count)
Welch Two Sample t-test
data: FirmSum[FirmSum$group == "1", ]$count and FirmSum[FirmSum$group == "2", ]$count
t = 3.0711, df = 14.745, p-value = 0.007889
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
533509 2965815
sample estimates:
mean of x mean of y
2650400.4 900738.4