Prophages of the Human Body
Bacteriophages (phages) are a ubiquitous part of all microbiomes and the human microbiome is no exception. Many spend time integrated into their host bacteria’s genomes as prophages, with almost all bacteria thought to have prophages. Prophages are often harmless to us but some can, while integrated into their hosts genomes, provide genes that protect the bacteria in various ways, including providing it with toxin-production genes. In these cases prophages can be a cause of illnesses such as cholera (cholera toxin), diphtheria (diphtheria toxin) or food poisoning (shiga toxin).
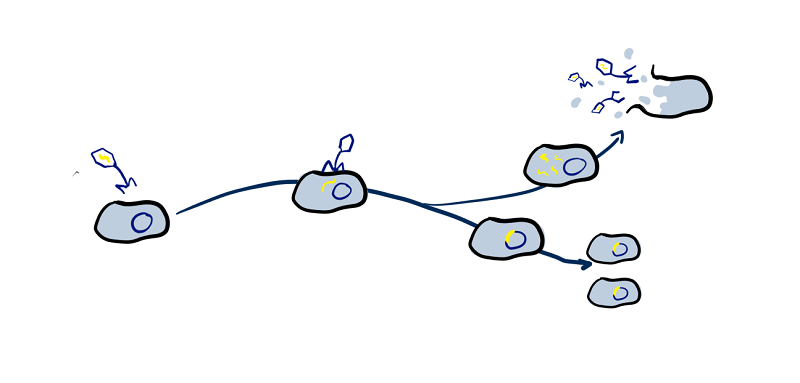
To gain an overview of just how common prophages are in the human microbiome we have mined GenBank for bacterial genomes isolated from different areas of the human body and analysed them with PhiSpy and AMRFinder+ to determine a) how common are prophages in the human microbiome and how much of the bacterial genomes are made up of prophage DNA, b) whether that amount of prophage DNA varies between areas of the body, or host health, and c) to find out how common phages that contain antimicrobial resistance and virulence genes are, what kinds of genes they can provide, and if there are any genes that are only provided by prophages.
The Human Panvirome
The human body contains a multitude of disparate microbial communities with viruses constituting a ubiquitous component of these microbiomes. Understanding viromes—the viral population of a microbiome—as a key component of human microbiomes is crucial to the future of human health. Unfortunately, this human meta-virome is currently poorly understood.
Hecatomb is a software pipeline developed in a collaboration between Flinders University, Washington University in Saint Louis, and San Diego State University to address many of the pitfalls of identifying viruses in metagenomes. Utilising HPC resources at Pawsey Supercomputing Research Centre, this project will apply Hecatomb, together with other phage tools such as Phables, Pharokka, and Phynteny to analyse tens of thousands of human metagenomics samples publicly available on the Sequence Read Archive (SRA) to create a human meta-virome.
Variation Analysis in Viral Metagenomes
Currently, our understanding of the scope of viral diversity in the environment is limited and there is an increasing demand for better computational tools to perform viral analysis. Constructing full-length viral genomes from high throughput sequencing data can be challenging due to the complex genome structures of viruses and most existing tools produce fragmented genomes. Viral identification tools rely on models trained using known genomes to predict if sequences originated from viral genomes and these tools may overlook novel viruses. Moreover, existing computational tools cannot identify viral genomes from environmental communities of similar viral variants. This project aims to address these shortcomings of existing methods and allow accurate recovery of complete and contiguous viral genomes that can facilitate downstream metagenomic analysis. We will propose new graph-based methods to recover and study complete viral genomes from metagenomic data without direct alignment to existing sequence databases. The recovered genomes will be analysed to study the structure and variation occurring within viral communities.

This project will apply Hecatomb, Phables, Pharokka and Phynteny to analyse and annotate viral genomes. Two types of analyses will be carried out to study the variation occurring in the recovered genomes from each viral component to understand the viral community dynamics.
- Graph-based analysis will be performed on identified viral components to capture different diversity metrics defining connectivity and dominance of recovered genomes.
- Gene-based analysis will be conducted on the resolved genomes to study the variation in genome structure and gene organisation.
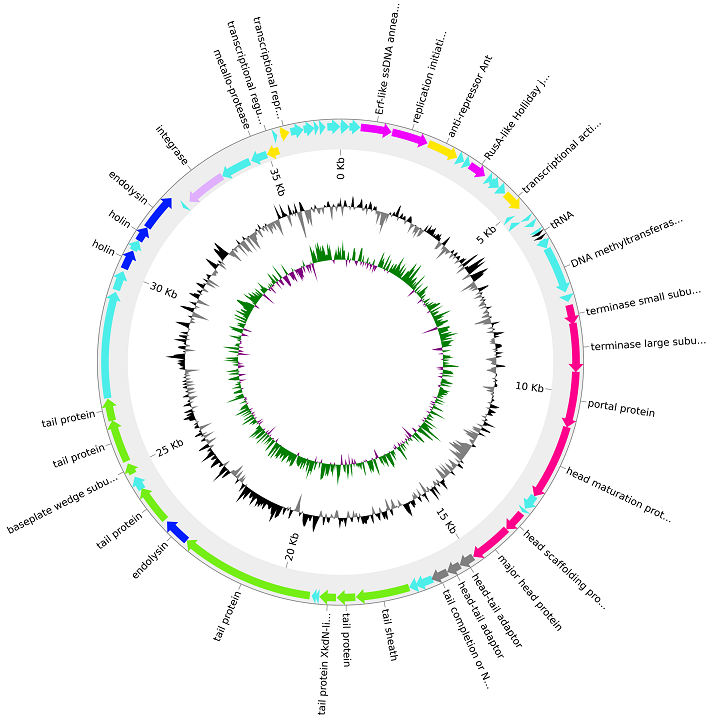
Profiling Prophages
Prophages are viruses that are integrated into bacterial genomes. Annotating prophages is extremely difficult, but we have built a FAIR framework for evaluating bioinformatics tools that attempt to do just this! Now published in F1000
Next, in the most comprehensive prophage analysis to date, we analysed over two and a half million prophages from over half a million bacterial genome assemblies. Analysis of the whole dataset and a representative subset of taxonomically diverse bacterial genomes demonstrated that the normalised prophage density was uniform across all bacterial genomes above 2 Mbp. Our prophage database is informing many of our ongoing exa-scale metagenomics projects. BioRxiv