
Deduplicating a genome assembly
Step 1: Login to Deepthought
You should have access via FLO with Jupyterhub: https://deepteachweb.flinders.edu.au/jupyter
Create a directory for this tutorial.
mkdir workshop
cd workshop/
Step 2: Download the files
For this we will be using the Clavicorona pyxidata assembly from the FALCON-Unzip paper: https://doi.org/10.1038/nmeth.4035 This is a small fungal genome, but, the overall process is exactly the same for deduplicating a plant genome.
We will need the sequencing reads. I have converted them to fasta format for you.
# Download the assembly
wget -O assembly.fasta.gz "https://cloudstor.aarnet.edu.au/plus/s/22dFsyUKMaPAldc/download"
# unzip the assembly
gunzip assembly.fasta.gz
# Download the PacBio longreads
wget -O reads.fasta.gz "https://cloudstor.aarnet.edu.au/plus/s/lKSei0Yn86eHVU0/download"
Step 3: Install the software
We will be using conda for all software. Create a new conda environment and attach to it.
conda create -n workshop
conda activate workshop
We need to install some software. We’ll use minimap2 and samtools for doing the alignments, quast for checking the assemblies, and purge haplotigs for deduplicating. minimap2 and samtools come with purge haplotigs, so the installation is simply:
# install quast
conda install -c conda-forge -c bioconda quast=5.2.0
# install purge haplotigs (plus minimap and samtools)
conda install -c conda-forge -c bioconda purge_haplotigs=1.1.2
Step 4: Check the draft assembly
Quast is a great program for quickly and easily getting the metrics for an assembly. Have a look at the documentation on GitHub: https://github.com/ablab/quast. There are few things you can do with Quast, but we just want to get the basic metrics for the assembly.
Run quast against the downloaded genome (try and figure out the command from the documentation)
Halp plz
quast assembly.fastaYou should now have a quast_results folder containing the quast report in a bunch of different formats. Download the report.html for a pretty report, or, cat the report.txt to look at the results in the teminal. We are expecting a 38 Mbp genome size.
cat quast_results/latest/report.txt
Assembly assembly
# contigs (>= 0 bp) 82
# contigs (>= 1000 bp) 82
# contigs (>= 5000 bp) 82
# contigs (>= 10000 bp) 82
# contigs (>= 25000 bp) 79
# contigs (>= 50000 bp) 62
Total length (>= 0 bp) 41902648
Total length (>= 1000 bp) 41902648
Total length (>= 5000 bp) 41902648
Total length (>= 10000 bp) 41902648
Total length (>= 25000 bp) 41830931
Total length (>= 50000 bp) 41211492
# contigs 82
Largest contig 4778091
Total length 41902648
GC (%) 54.43
N50 1484160
N90 252656
auN 1860737.6
L50 8
L90 36
# N's per 100 kbp 0.00
Is the total length what we are expecting? Do we need to deduplicate?
Hint: yes we do!
Step 5: Align the reads to the assembly
Before running Purge Haplotigs we need to map the reads to the assembly. This is to identify the contigs with half-coverage and mark them as potential duplicates. Have a look at the Purge Haplotigs documentation: https://bitbucket.org/mroachawri/purge_haplotigs/src/master/
Align the reads with minimap2 and pipe to samtools sort to create a sorted BAM file
Again, try to figure out the command that you need to run.
Halp plz
minimap2 -ax map-pb assembly.fasta reads.fasta.gz | samtools sort > alignment.bamIs it running too slowly? Download the alignment.
wget -O aligned.bam "https://cloudstor.aarnet.edu.au/plus/s/gJdHF2frAZjXocp/download"
Step 6: Generate a read-depth histogram
The first step of Purge Haplotigs is to generate a read-depth histogram.
For this we use purge_haplotigs hist.
Run this step. You should have two files: aligned.bam.histogram.png which is the histogram,
and aligned.bam.gencov which has the read depth for all contigs.
Download aligned.bam.histogram.png and have a look at the peaks.
You should see two peaks, with the smaller peak corresponding to duplicated contigs,
and the larger peak corresponding to the haplotype-fused contigs.
Halp plz
purge_haplotigs hist -b alignment.bam -g assembly.fastaPick some cutoffs to capture the two peaks
You need a low cutoff -l, a midpoint -m, and a high cutoff -h.
Check out this example to give you an idea exampleHistogram.png
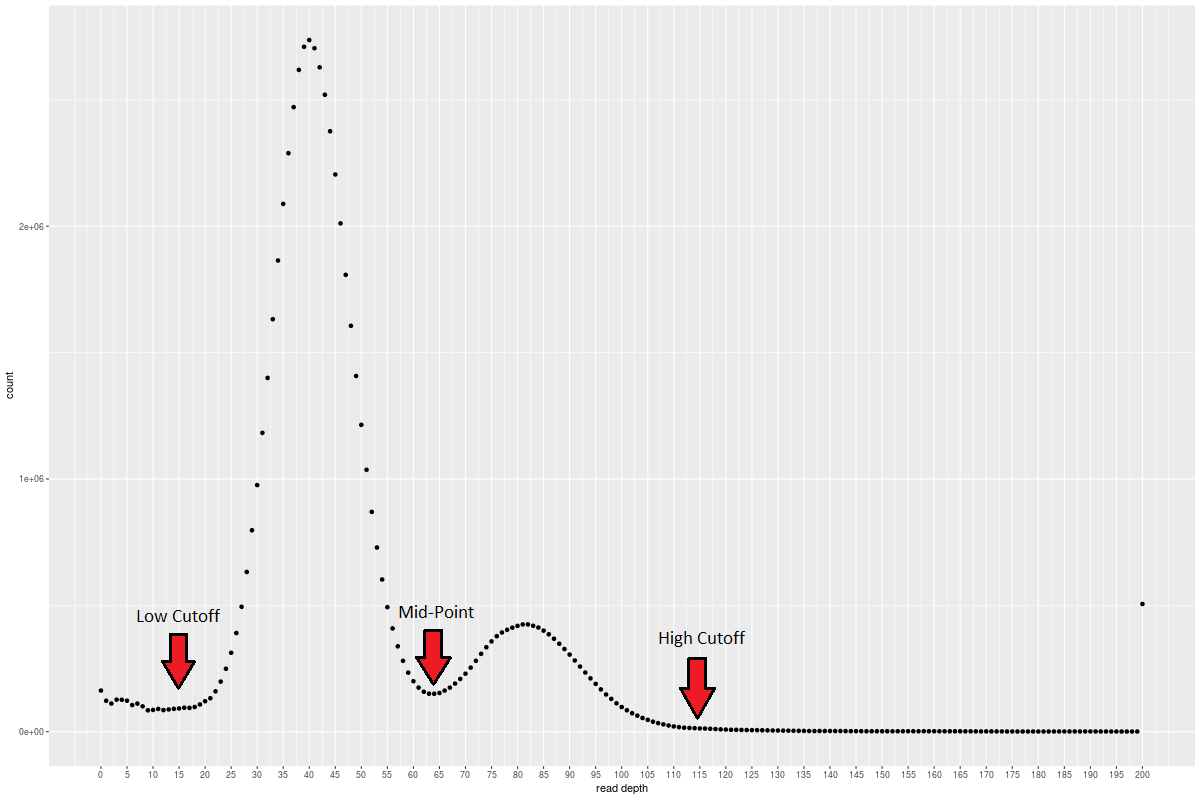{kind=link}
Halp plz
I'm going with low=10, mid=40, high=110Step 7: Mark potential duplicate contigs
You’ll now run purge_haplotigs cov with your selected cutoffs to mark the potential duplicates.
This step will create a coverage_stats.csv which will have the “junk” contigs and suspected duplicates marked.
Halp plz
purge_haplotigs cov -i aligned.bam.gencov -l 15 -m 40 -h 110Step 8: Purge the duplicate contigs
This step will compare the marked contigs with each other and identify true duplicates.
You’ll get a curated.fasta file which will be the new assembly and the curated.haplotigs.fasta file
which will contain the purged duplicates.
Halp plz
purge_haplotigs purge -g assembly.fasta -c coverage_stats.csvStep 9: Re-evaluate the assembly
Let’s see how our assembly compares to before. Rerun Quast and see if we’re closer to our expected genome size of 38 Mbp.
Halp plz
quast curated.fastaYou should get something like this:
Assembly curated
# contigs (>= 0 bp) 49
# contigs (>= 1000 bp) 49
# contigs (>= 5000 bp) 49
# contigs (>= 10000 bp) 49
# contigs (>= 25000 bp) 49
# contigs (>= 50000 bp) 46
Total length (>= 0 bp) 38966126
Total length (>= 1000 bp) 38966126
Total length (>= 5000 bp) 38966126
Total length (>= 10000 bp) 38966126
Total length (>= 25000 bp) 38966126
Total length (>= 50000 bp) 38835102
# contigs 49
Largest contig 4778091
Total length 38966126
GC (%) 54.66
N50 1586845
N90 402215
auN 1986847.2
L50 7
L90 27
# N's per 100 kbp 0.00
Much better!
If you’re bored, you could remap the reads to the new assembly and rerun purge_haplotigs hist
to see how the bimodal peaks compare to before.
Congrats!
You’ve just deduplicated an assembly. yay!
Bonus round
Download the original Haplotigs.
wget -O assembly.haplotigs.fasta "https://cloudstor.aarnet.edu.au/plus/s/nOWmpNiemvUUsjQ/download"
Combine them with the reassigned haplotigs and create a dotplot with MUMmer.